A "Best of the Best Practices" (BOBP) guide to developing in Python.
- "Build tools for others that you want to be built for you." - Kenneth Reitz
- "Simplicity is alway better than functionality." - Pieter Hintjens
| UPSTREAM=400 | |
| INSTREAM=100 | |
| ORG=hg18 | |
| mysql --user genome --host genome-mysql.cse.ucsc.edu -NAD $ORG -e \ | |
| "select chrom, txStart, txEnd, X.geneSymbol, strand from knownGene as K, kgXref as X WHERE txStart != txEnd AND X.kgID = K.name" \ | |
| | awk -v ups=$UPSTREAM -v ins=$INSTREAM 'BEGIN{OFS=FS="\t"} | |
| $5 == "-" { print $1,$3-ins,$3+ups,$4 } | |
| $5 == "+" { print $1,$2-ins,$2+ups,$4 }' \ | |
| | sort -k1,1 -k2,2n \ |
| # Palette from: http://flatuicolors.com/ | |
| # Usage: | |
| # library(devtools) | |
| # devtools::source_gist('0aebc4c2fe2f4298065b') | |
| teal = rgb(26, 188, 156, maxColorValue= 255) | |
| dark_teal = rgb(22, 160, 133, maxColorValue=255) | |
| green = rgb(46, 204, 113, maxColorValue=255) | |
| dark_green = rgb(39, 174, 96, maxColorValue=255) | |
| blue = rgb(52, 152, 219, maxColorValue=255) | |
| dark_blue = rgb(41, 128, 185, maxColorValue=255) |
| #' Supplementary Data 2 | |
| #' ======================= | |
| #' ### Supplementary code and figures accompanying *Interconnections among mutations, gene expression, clinical variables and patient outcome in myelodysplastic syndromes* | |
| #' | |
| #' This document contains the complete code used in the analysis. It is purely written in `R` using a series of `R` and `Bioconductor` packages. | |
| #' This report has been generated using the `knitr` R package (http://yihui.name/knitr/). | |
| #' For a complete list of packages and their versions please have a look at the end of this document. | |
| #+ options, echo=FALSE, eval=TRUE | |
| options(width=120) |
This hit #rstats today:
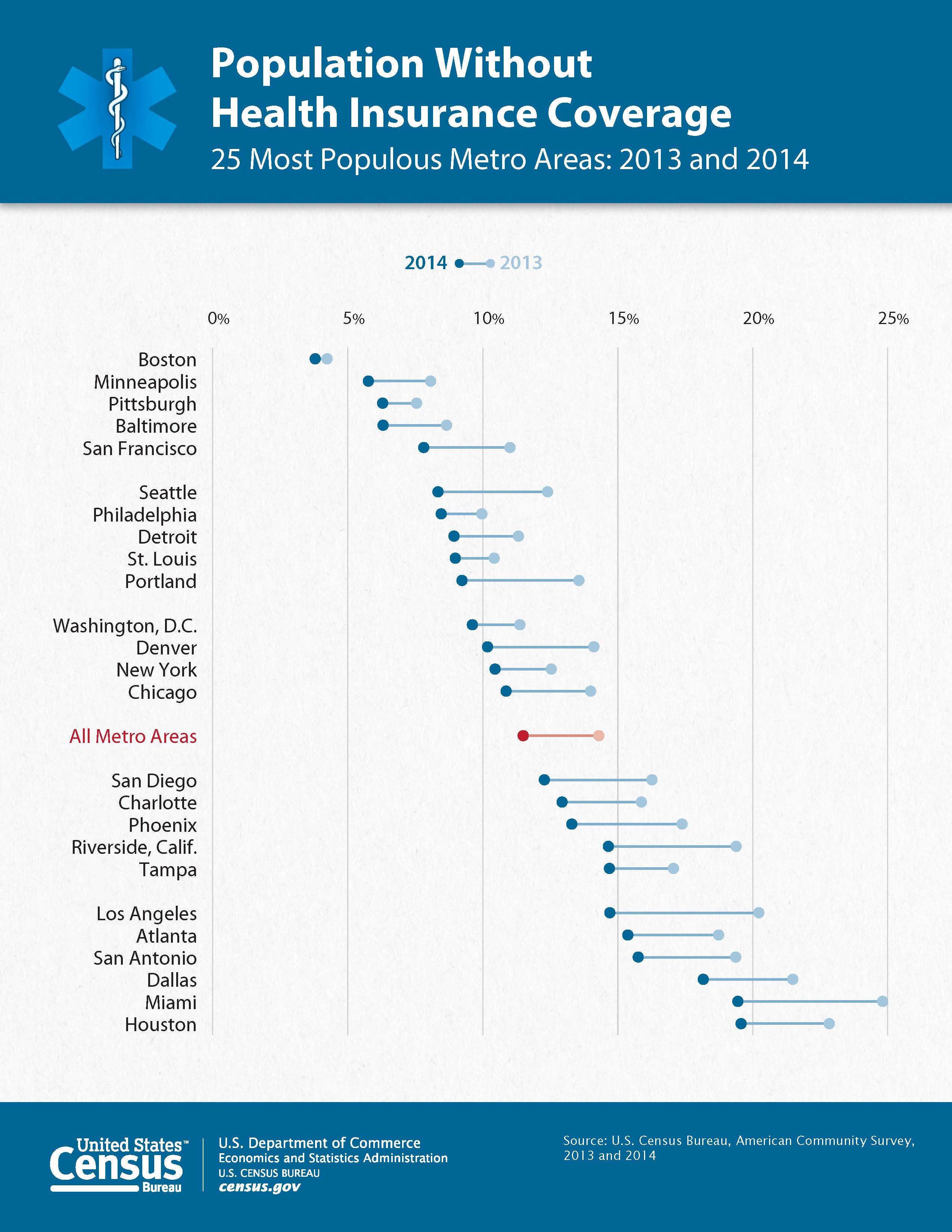
Has anyone made a dumbbell dot plot in #rstats, or better yet exported to @plotlygraphs using the API? https://t.co/rWUSpH1rRl
— Ken Davis (@ken_mke) October 23, 2015So, I figured it was worth a cpl mins to reproduce.
While the US gov did give the data behind the chart it was all the data and a pain to work with so I used WebPlotDigitizer to transcribe the points and then some data wrangling in R to clean it up and make it work well with ggplot2.
It is possible to make the top "dumbbell" legend in ggplot2 (but not by using a guide) and color the "All Metro A
Sometimes you may want to undo a whole commit with all changes. Instead of going through all the changes manually, you can simply tell git to revert a commit, which does not even have to be the last one. Reverting a commit means to create a new commit that undoes all changes that were made in the bad commit. Just like above, the bad commit remains there, but it no longer affects the the current master and any future commits on top of it.
git revert {commit_id}
Deleting the last commit is the easiest case. Let's say we have a remote origin with branch master that currently points to commit dd61ab32. We want to remove the top commit. Translated to git terminology, we want to force the master branch of the origin remote repository to the parent of dd61ab32:
| ## Purpose: illustrate use of magick, gganimate, and purrr | |
| ## Inspiration: | |
| ## https://rud.is/b/2016/07/27/u-s-drought-animations-with-the-witchs-brew-purrr-broom-magick/ | |
| ## and subsequent discussion on magick vs gganimate: | |
| ## https://twitter.com/hrbrmstr/status/758304420224466944 | |
| library(purrr) | |
| library(ggplot2) | |
| library(gganimate) | |
| library(animation) | |
| library(magick) |
| first_reads=$1 | |
| second_reads=$2 | |
| ddir=$(dirname $first_reads) | |
| obase_first=$(basename $first_reads .fastq.gz) | |
| obase_second=$(basename $second_reads .fastq.gz) | |
| splitsz=4000000 | |
| if [ ! -z ${first_reads} ] && [ -e ${first_reads} ] |
| *bcftools filter | |
| *Filter variants per region (in this example, print out only variants mapped to chr1 and chr2) | |
| qbcftools filter -r1,2 ALL.chip.omni_broad_sanger_combined.20140818.snps.genotypes.hg38.vcf.gz | |
| *printing out info for only 2 samples: | |
| bcftools view -s NA20818,NA20819 filename.vcf.gz | |
| *printing stats only for variants passing the filter: | |
| bcftools view -f PASS filename.vcf.gz |
{kind=link}