Created
April 26, 2017 19:27
-
-
Save sbliven/b7cc2c5305aa3652a75a580ae7c6ce33 to your computer and use it in GitHub Desktop.
Convert mmCIF files to PDB format using biopython
This file contains hidden or bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| #!/usr/bin/env python | |
| """ | |
| Script to convert mmCIF files to PDB format. | |
| usage: python cif2pdb.py ciffile [pdbfile] | |
| Requires python BioPython (`pip install biopython`). It should work with recent version of python 2 or 3. | |
| @author Spencer Bliven <[email protected]> | |
| """ | |
| import sys | |
| import argparse | |
| import logging | |
| from Bio.PDB.MMCIFParser import MMCIFParser | |
| from Bio.PDB import PDBIO | |
| def int_to_chain(i,base=62): | |
| """ | |
| int_to_chain(int,int) -> str | |
| Converts a positive integer to a chain ID. Chain IDs include uppercase | |
| characters, numbers, and optionally lowercase letters. | |
| i = a positive integer to convert | |
| base = the alphabet size to include. Typically 36 or 62. | |
| """ | |
| if i < 0: | |
| raise ValueError("positive integers only") | |
| if base < 0 or 62 < base: | |
| raise ValueError("Invalid base") | |
| quot = int(i)//base | |
| rem = i%base | |
| if rem < 26: | |
| letter = chr( ord("A") + rem) | |
| elif rem < 36: | |
| letter = str( rem-26) | |
| else: | |
| letter = chr( ord("a") + rem - 36) | |
| if quot == 0: | |
| return letter | |
| else: | |
| return int_to_chain(quot-1,base) + letter | |
| class OutOfChainsError(Exception): pass | |
| def rename_chains(structure): | |
| """Renames chains to be one-letter chains | |
| Existing one-letter chains will be kept. Multi-letter chains will be truncated | |
| or renamed to the next available letter of the alphabet. | |
| If more than 62 chains are present in the structure, raises an OutOfChainsError | |
| Returns a map between new and old chain IDs, as well as modifying the input structure | |
| """ | |
| next_chain = 0 # | |
| # single-letters stay the same | |
| chainmap = {c.id:c.id for c in structure.get_chains() if len(c.id) == 1} | |
| for o in structure.get_chains(): | |
| if len(o.id) != 1: | |
| if o.id[0] not in chainmap: | |
| chainmap[o.id[0]] = o.id | |
| o.id = o.id[0] | |
| else: | |
| c = int_to_chain(next_chain) | |
| while c in chainmap: | |
| next_chain += 1 | |
| c = int_to_chain(next_chain) | |
| if next_chain >= 62: | |
| raise OutOfChainsError() | |
| chainmap[c] = o.id | |
| o.id = c | |
| return chainmap | |
| if __name__ == "__main__": | |
| parser = argparse.ArgumentParser(description='Convert mmCIF to PDB format') | |
| parser.add_argument("ciffile",help="mmCIF input file") | |
| parser.add_argument("pdbfile",nargs="?",help="PDB output file. Default based on CIF filename") | |
| parser.add_argument("-v","--verbose", help="Long messages", | |
| dest="verbose",default=False, action="store_true") | |
| args = parser.parse_args() | |
| logging.basicConfig(format='%(levelname)s: %(message)s', level=logging.DEBUG if args.verbose else logging.WARN) | |
| ciffile = args.ciffile | |
| pdbfile = args.pdbfile or ciffile+".pdb" | |
| #Not sure why biopython needs this to read a cif file | |
| strucid = ciffile[:4] if len(ciffile)>4 else "1xxx" | |
| # Read file | |
| parser = MMCIFParser() | |
| structure = parser.get_structure(strucid, ciffile) | |
| # rename long chains | |
| try: | |
| chainmap = rename_chains(structure) | |
| except OutOfChainsError: | |
| logging.error("Too many chains to represent in PDB format") | |
| sys.exit(1) | |
| if args.verbose: | |
| for new,old in chainmap.items(): | |
| if new != old: | |
| logging.info("Renaming chain {0} to {1}".format(old,new)) | |
| #Write PDB | |
| io = PDBIO() | |
| io.set_structure(structure) | |
| #TODO What happens with large structures? | |
| io.save(pdbfile) | |
Sign up for free
to join this conversation on GitHub.
Already have an account?
Sign in to comment
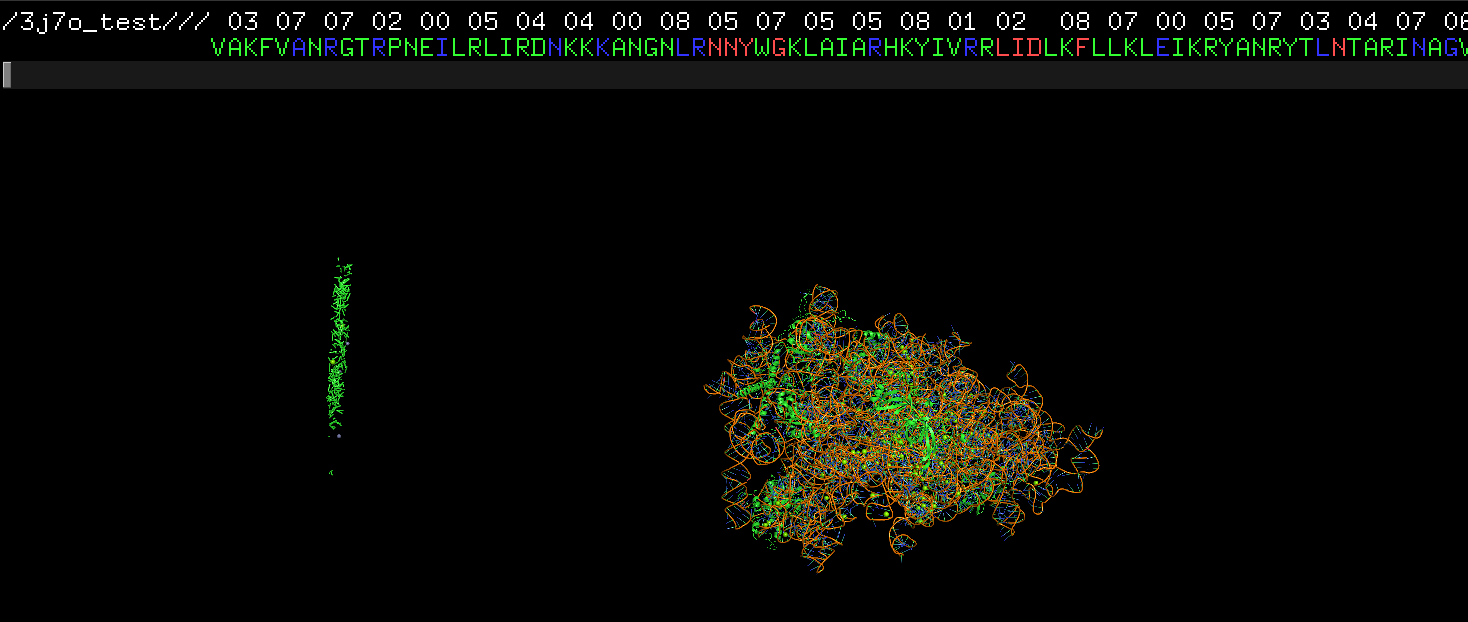
Hi, I have question regarding the result. As you can see from the screenshot attached, there are some dots/lines of amino acids far away from the main protein structure. This is the pdb file after cif2pdb.py `for` 3j7o.cif. Those seem like some abandoned amino acid. Do you have any idea why that happend? I also got some warning messages:/homes/prod/anaconda3/envs/ngs/lib/python3.9/site-packages/biopython-1.79-py3.9-linux-x86_64.egg/Bio/PDB/StructureBuilder.py:89: PDBConstructionWarning: WARNING: Chain 5 is discontinuous at line 136682. warnings.warn( /homes/prod/anaconda3/envs/ngs/lib/python3.9/site-packages/biopython-1.79-py3.9-linux-x86_64.egg/Bio/PDB/StructureBuilder.py:89: PDBConstructionWarning: WARNING: Chain 7 is discontinuous at line 136801. warnings.warn( /homes/prod/anaconda3/envs/ngs/lib/python3.9/site-packages/biopython-1.79-py3.9-linux-x86_64.egg/Bio/PDB/StructureBuilder.py:89: PDBConstructionWarning: WARNING: Chain 8 is discontinuous at line 136806. warnings.warn( /homes/prod/anaconda3/envs/ngs/lib/python3.9/site-packages/biopython-1.79-py3.9-linux-x86_64.egg/Bio/PDB/StructureBuilder.py:89: PDBConstructionWarning: WARNING: Chain P is discontinuous at line 136810.Thanks for the help!