-
-
Save slowkow/52a8911e18a873a51dcb to your computer and use it in GitHub Desktop.
| #!/usr/bin/env bash | |
| # reads_per_seq | |
| # | |
| # Run 8 jobs in parallel on many BAM files: | |
| # | |
| # parallel -j8 reads_per_seq ::: *.bam | |
| set -eou pipefail | |
| if [[ $# -ne 1 || "$1" != *.bam ]]; then | |
| echo "usage: $(basename $0) file.bam" | |
| echo | |
| echo " Produce a tab-delimited file.seq with number per sequence of" | |
| echo " primary alignments with proper read pairs." | |
| echo | |
| exit 1 | |
| fi | |
| bam="$1" | |
| # Read more about samtools options: http://www.htslib.org/doc/samtools.html | |
| # | |
| # Learn more about samtools flags: https://www.samformat.info/sam-format-flag | |
| # | |
| samtools view -f 2 -F 104 "$bam" \ | |
| | cut -f3 | sort | uniq -c | sort -k1nr \ | |
| | awk 'OFS="\t" {print $1,$2}' \ | |
| > "${bam%.bam}.seq" |
Thanks!
Indeed, it's important to use one of those tools to figure out which decimal you should use.
I like this one:
https://www.samformat.info/sam-format-flag
I think the comments in the script may be misleading because showing octal instead of decimal might tempt the reader to add them up like decimal numbers. Sorry about that!
At the time of writing (2015), I think I wanted a count, for each chromosome, of "the number of primary alignments with proper read pairs".
Which do you think is the right decimal for that goal? My understanding is -F DECIMAL means that samtools will not count an alignment that matches any of those flags (in other words, it will use the Boolean OR operator).
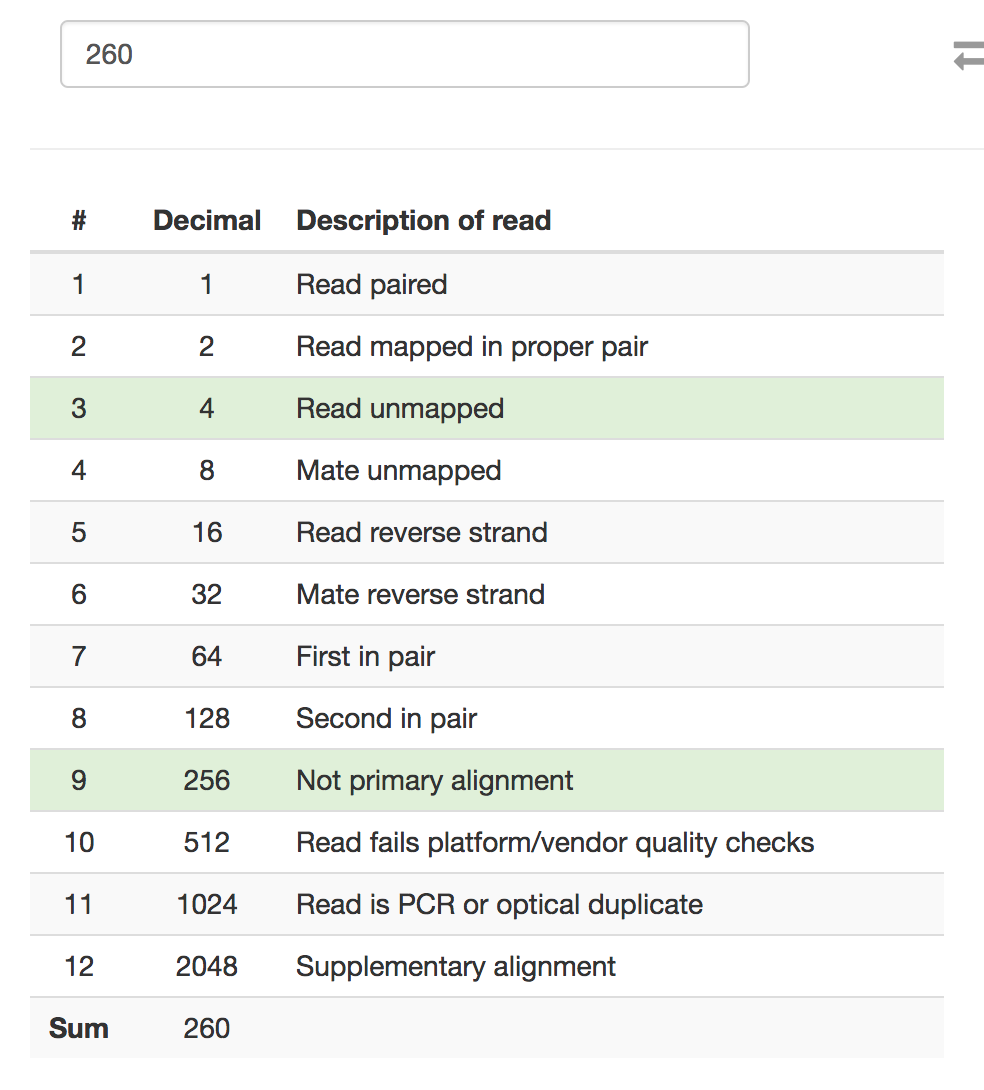
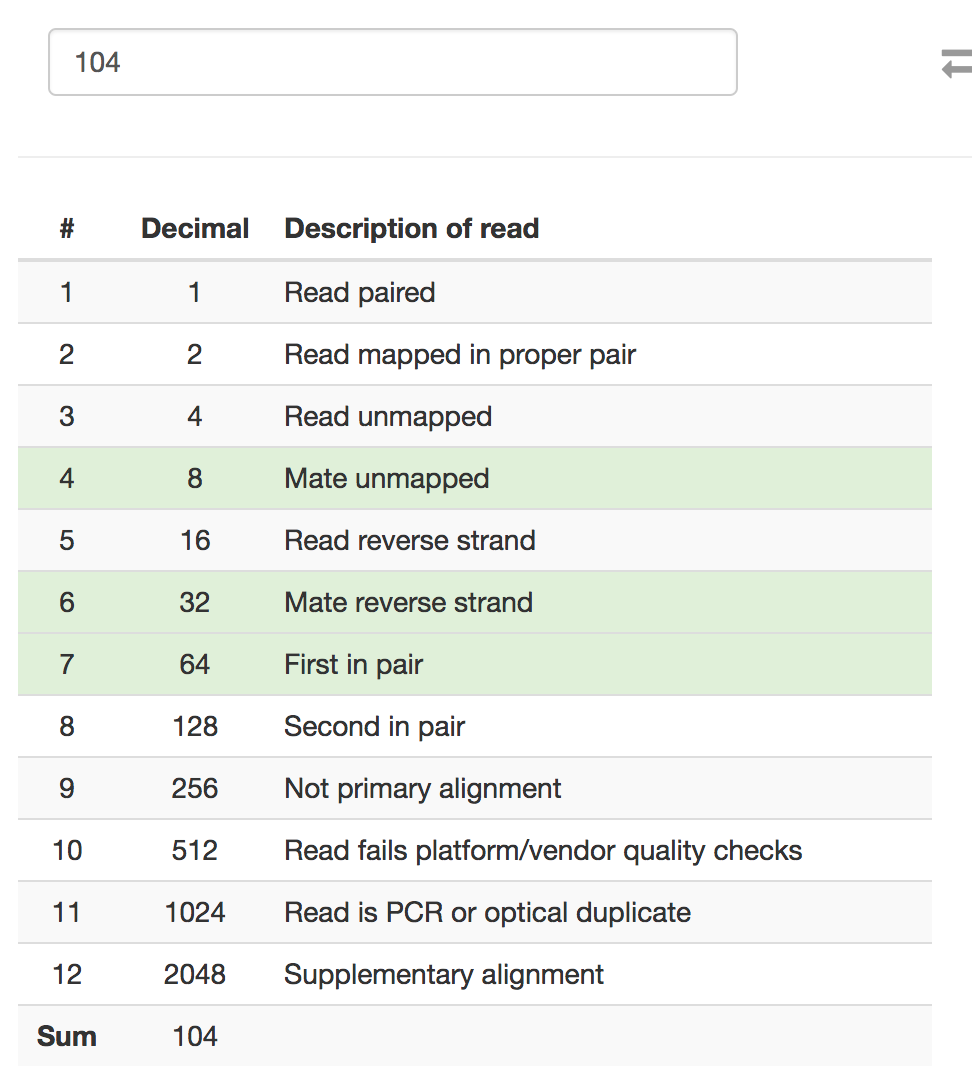
If counting alignments in BAMs is important for your work, then I would recommend doing some experiments on the command line to figure out which flags are giving you the results you would expect to see. Better to verify than to trust what you find online.
You might want to check the
-F 104portion of the samtools command. Based on the Picard SAM FLAG convertor, "fragment unmapped" + "secondary alignment" is 260 in decimal, not 104.